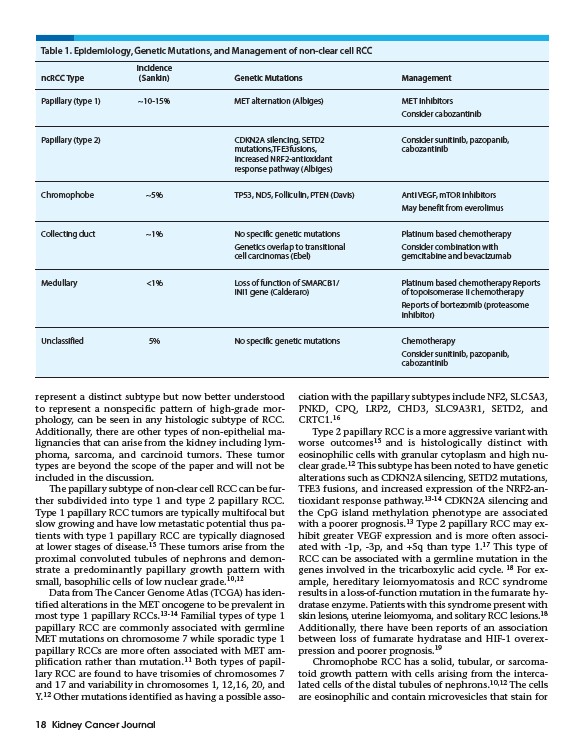
Table 1. Epidemiology, Genetic Mutations, and Management of non-clear cell RCC
represent a distinct subtype but now better understood
to represent a nonspecific pattern of high-grade morphology,
can be seen in any histologic subtype of RCC.
Additionally, there are other types of non-epithelial malignancies
that can arise from the kidney including lymphoma,
sarcoma, and carcinoid tumors. These tumor
types are beyond the scope of the paper and will not be
included in the discussion.
The papillary subtype of non-clear cell RCC can be further
subdivided into type 1 and type 2 papillary RCC.
Type 1 papillary RCC tumors are typically multifocal but
slow growing and have low metastatic potential thus patients
with type 1 papillary RCC are typically diagnosed
at lower stages of disease.15 These tumors arise from the
proximal convoluted tubules of nephrons and demonstrate
a predominantly papillary growth pattern with
small, basophilic cells of low nuclear grade.10,12
Data from The Cancer Genome Atlas (TCGA) has identified
alterations in the MET oncogene to be prevalent in
most type 1 papillary RCCs.13-14 Familial types of type 1
papillary RCC are commonly associated with germline
MET mutations on chromosome 7 while sporadic type 1
papillary RCCs are more often associated with MET amplification
rather than mutation.11 Both types of papillary
RCC are found to have trisomies of chromosomes 7
and 17 and variability in chromosomes 1, 12,16, 20, and
Y.12 Other mutations identified as having a possible association
18 Kidney Cancer Journal
with the papillary subtypes include NF2, SLC5A3,
PNKD, CPQ, LRP2, CHD3, SLC9A3R1, SETD2, and
CRTC1.16
Type 2 papillary RCC is a more aggressive variant with
worse outcomes15 and is histologically distinct with
eosinophilic cells with granular cytoplasm and high nuclear
grade.12 This subtype has been noted to have genetic
alterations such as CDKN2A silencing, SETD2 mutations,
TFE3 fusions, and increased expression of the NRF2-antioxidant
response pathway.13-14 CDKN2A silencing and
the CpG island methylation phenotype are associated
with a poorer prognosis.13 Type 2 papillary RCC may exhibit
greater VEGF expression and is more often associated
with -1p, -3p, and +5q than type 1.17 This type of
RCC can be associated with a germline mutation in the
genes involved in the tricarboxylic acid cycle. 18 For example,
hereditary leiomyomatosis and RCC syndrome
results in a loss-of-function mutation in the fumarate hydratase
enzyme. Patients with this syndrome present with
skin lesions, uterine leiomyoma, and solitary RCC lesions.18
Additionally, there have been reports of an association
between loss of fumarate hydratase and HIF-1 overexpression
and poorer prognosis.19
Chromophobe RCC has a solid, tubular, or sarcomatoid
growth pattern with cells arising from the intercalated
cells of the distal tubules of nephrons.10,12 The cells
are eosinophilic and contain microvesicles that stain for
Incidence
ncRCC Type (Sankin) Genetic Mutations Management
Papillary (type 1) ~10-15% MET alternation (Albiges) MET inhibitors
Consider cabozantinib
Papillary (type 2) CDKN2A silencing, SETD2 Consider sunitinib, pazopanib,
mutations,TFE3fusions, cabozantinib
increased NRF2-antioxidant
response pathway (Albiges)
Chromophobe ~5% TP53, ND5, Folliculin, PTEN (Davis) Anti VEGF, mTOR inhibitors
May benefit from everolimus
Collecting duct ~1% No specific genetic mutations Platinum based chemotherapy
Genetics overlap to transitional Consider combination with
cell carcinomas (Ebel) gemcitabine and bevacizumab
Medullary <1% Loss of function of SMARCB1/ Platinum based chemotherapy Reports
INI1 gene (Calderaro) of topoisomerase II chemotherapy
Reports of bortezomib (proteasome
inhibitor)
Unclassified 5% No specific genetic mutations Chemotherapy
Consider sunitinib, pazopanib,
cabozantinib