Kidney Cancer Journal 95
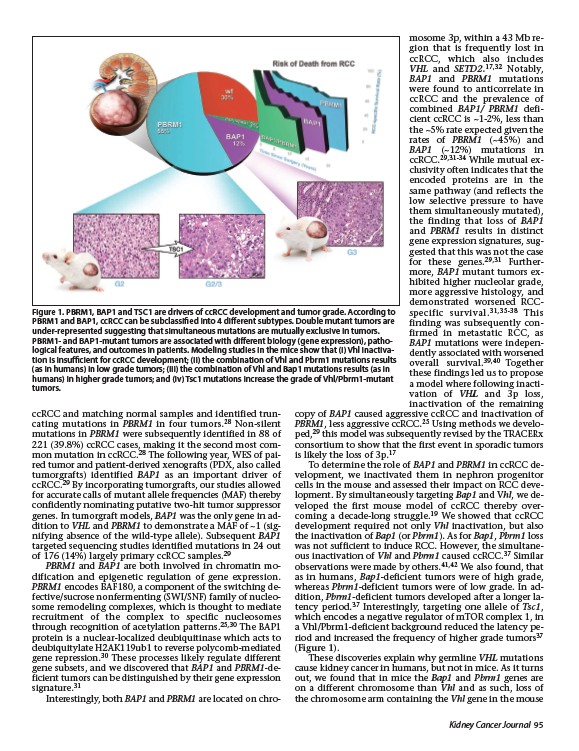
Figure 1. PBRM1, BAP1 and TSC1 are drivers of ccRCC development and tumor grade. According to
PBRM1 and BAP1, ccRCC can be subclassified into 4 different subtypes. Double mutant tumors are
under-represented suggesting that simultaneous mutations are mutually exclusive in tumors.
PBRM1- and BAP1-mutant tumors are associated with different biology (gene expression), pathological
features, and outcomes in patients. Modeling studies in the mice show that (i) Vhl inactivation
is insufficient for ccRCC development; (ii) the combination of Vhl and Pbrm1 mutations results
(as in humans) in low grade tumors; (iii) the combination of Vhl and Bap1 mutations results (as in
humans) in higher grade tumors; and (iv) Tsc1 mutations increase the grade of Vhl/Pbrm1-mutant
tumors.
ccRCC and matching normal samples and identified truncating
mutations in PBRM1 in four tumors.28 Non-silent
mutations in PBRM1 were subsequently identified in 88 of
221 (39.8%) ccRCC cases, making it the second most common
mutation in ccRCC.28 The following year, WES of paired
tumor and patient-derived xenografts (PDX, also called
tumorgrafts) identified BAP1 as an important driver of
ccRCC.29 By incorporating tumorgrafts, our studies allowed
for accurate calls of mutant allele frequencies (MAF) thereby
confidently nominating putative two-hit tumor suppressor
genes. In tumorgraft models, BAP1 was the only gene in addition
to VHL and PBRM1 to demonstrate a MAF of ~1 (signifying
absence of the wild-type allele). Subsequent BAP1
targeted sequencing studies identified mutations in 24 out
of 176 (14%) largely primary ccRCC samples.29
PBRM1 and BAP1 are both involved in chromatin modification
and epigenetic regulation of gene expression.
PBRM1 encodes BAF180, a component of the switching defective/
sucrose nonfermenting (SWI/SNF) family of nucleosome
remodeling complexes, which is thought to mediate
recruitment of the complex to specific nucleosomes
through recognition of acetylation patterns.25,30 The BAP1
protein is a nuclear-localized deubiquitinase which acts to
deubiquitylate H2AK119ub1 to reverse polycomb-mediated
gene repression.30 These processes likely regulate different
gene subsets, and we discovered that BAP1 and PBRM1-deficient
tumors can be distinguished by their gene expression
signature.31
Interestingly, both BAP1 and PBRM1 are located on chromosome
3p, within a 43 Mb region
that is frequently lost in
ccRCC, which also includes
VHL and SETD2.17,32 Notably,
BAP1 and PBRM1 mutations
were found to anticorrelate in
ccRCC and the prevalence of
combined BAP1/ PBRM1 deficient
ccRCC is ~1-2%, less than
the ~5% rate expected given the
rates of PBRM1 (~45%) and
BAP1 (~12%) mutations in
ccRCC.29,31-34 While mutual exclusivity
often indicates that the
encoded proteins are in the
same pathway (and reflects the
low selective pressure to have
them simultaneously mutated),
the finding that loss of BAP1
and PBRM1 results in distinct
gene expression signatures, suggested
that this was not the case
for these genes.29,31 Furthermore,
BAP1 mutant tumors exhibited
higher nucleolar grade,
more aggressive histology, and
demonstrated worsened RCCspecific
survival.31,35-38 This
finding was subsequently confirmed
in metastatic RCC, as
BAP1 mutations were independently
associated with worsened
overall survival.39,40 Together
these findings led us to propose
a model where following inactivation
of VHL and 3p loss,
inactivation of the remaining
copy of BAP1 caused aggressive ccRCC and inactivation of
PBRM1, less aggressive ccRCC.25 Using methods we developed,
29 this model was subsequently revised by the TRACERx
consortium to show that the first event in sporadic tumors
is likely the loss of 3p.17
To determine the role of BAP1 and PBRM1 in ccRCC development,
we inactivated them in nephron progenitor
cells in the mouse and assessed their impact on RCC development.
By simultaneously targeting Bap1 and Vhl, we developed
the first mouse model of ccRCC thereby over-
coming a decade-long struggle.19 We showed that ccRCC
development required not only Vhl inactivation, but also
the inactivation of Bap1 (or Pbrm1). As for Bap1, Pbrm1 loss
was not sufficient to induce RCC. However, the simultaneous
inactivation of Vhl and Pbrm1 caused ccRCC.37 Similar
observations were made by others.41,42 We also found, that
as in humans, Bap1-deficient tumors were of high grade,
whereas Pbrm1-deficient tumors were of low grade. In addition,
Pbrm1-deficient tumors developed after a longer latency
period.37 Interestingly, targeting one allele of Tsc1,
which encodes a negative regulator of mTOR complex 1, in
a Vhl/Pbrm1-deficient background reduced the latency period
and increased the frequency of higher grade tumors37
(Figure 1).
These discoveries explain why germline VHL mutations
cause kidney cancer in humans, but not in mice. As it turns
out, we found that in mice the Bap1 and Pbrm1 genes are
on a different chromosome than Vhl and as such, loss of
the chromosome arm containing the Vhl gene in the mouse