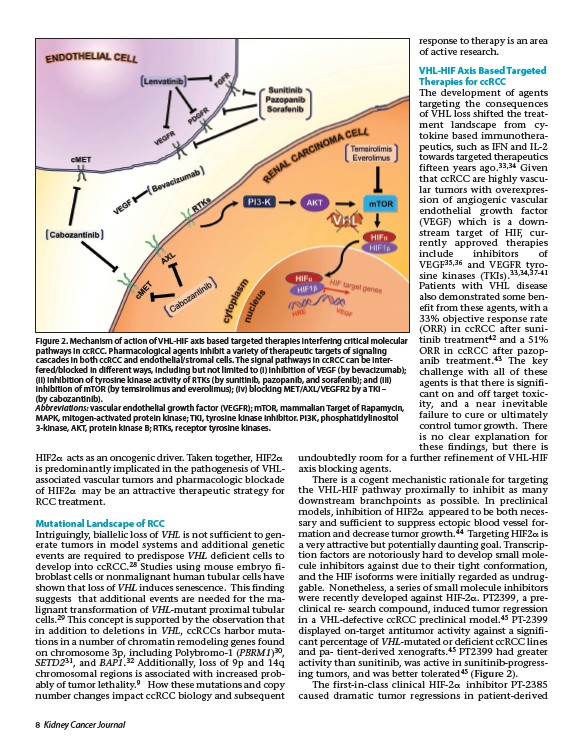
Figure 2. Mechanism of action of VHL-HIF axis based targeted therapies interfering critical molecular
pathways in ccRCC. Pharmacological agents inhibit a variety of therapeutic targets of signaling
cascades in both ccRCC and endothelial/stromal cells. The signal pathways in ccRCC can be interfered/
blocked in different ways, including but not limited to (i) inhibition of VEGF (by bevacizumab);
(ii) inhibition of tyrosine kinase activity of RTKs (by sunitinib, pazopanib, and sorafenib); and (iii)
inhibition of mTOR (by temsirolimus and everolimus); (iv) blocking MET/AXL/VEGFR2 by a TKI –
(by cabozantinib).
Abbreviations: vascular endothelial growth factor (VEGFR); mTOR, mammalian Target of Rapamycin,
MAPK, mitogen-activated protein kinase; TKI, tyrosine kinase inhibitor. PI3K, phosphatidylinositol
3-kinase, AKT, protein kinase B; RTKs, receptor tyrosine kinases.
HIF2a acts as an oncogenic driver. Taken together, HIF2a
is predominantly implicated in the pathogenesis of VHLassociated
vascular tumors and pharmacologic blockade
of HIF2a may be an attractive therapeutic strategy for
RCC treatment.
Mutational Landscape of RCC
Intriguingly, biallelic loss of VHL is not sufficient to generate
tumors in model systems and additional genetic
events are required to predispose VHL deficient cells to
develop into ccRCC.28 Studies using mouse embryo fibroblast
cells or nonmalignant human tubular cells have
shown that loss of VHL induces senescence. This finding
suggests that additional events are needed for the malignant
transformation of VHL-mutant proximal tubular
cells.29 This concept is supported by the observation that
in addition to deletions in VHL, ccRCCs harbor mutations
in a number of chromatin remodeling genes found
on chromosome 3p, including Polybromo-1 (PBRM1)30,
SETD231, and BAP1.32 Additionally, loss of 9p and 14q
chromosomal regions is associated with increased probably
of tumor lethality.9 How these mutations and copy
number changes impact ccRCC biology and subsequent
8 Kidney Cancer Journal
response to therapy is an area
of active research.
VHL-HIF Axis Based Targeted
Therapies for ccRCC
The development of agents
targeting the consequences
of VHL loss shifted the treatment
landscape from cytokine
based immunothera-
peutics, such as IFN and IL-2
towards targeted therapeutics
fifteen years ago.33,34 Given
that ccRCC are highly vascular
tumors with overexpression
of angiogenic vascular
endothelial growth factor
(VEGF) which is a downstream
target of HIF, currently
approved therapies
include inhibitors of
VEGF35,36 and VEGFR tyrosine
kinases (TKIs).33,34,37-41
Patients with VHL disease
also demonstrated some benefit
from these agents, with a
33% objective response rate
(ORR) in ccRCC after sunitinib
treatment42 and a 51%
ORR in ccRCC after pazopanib
treatment.43 The key
challenge with all of these
agents is that there is significant
on and off target toxicity,
and a near inevitable
failure to cure or ultimately
control tumor growth. There
is no clear explanation for
these findings, but there is
undoubtedly room for a further refinement of VHL-HIF
axis blocking agents.
There is a cogent mechanistic rationale for targeting
the VHL-HIF pathway proximally to inhibit as many
downstream branchpoints as possible. In preclinical
models, inhibition of HIF2a appeared to be both necessary
and sufficient to suppress ectopic blood vessel formation
and decrease tumor growth.44 Targeting HIF2a is
a very attractive but potentially daunting goal. Transcription
factors are notoriously hard to develop small molecule
inhibitors against due to their tight conformation,
and the HIF isoforms were initially regarded as undruggable.
Nonetheless, a series of small molecule inhibitors
were recently developed against HIF-2a. PT2399, a preclinical
re- search compound, induced tumor regression
in a VHL-defective ccRCC preclinical model.45 PT-2399
displayed on-target antitumor activity against a significant
percentage of VHL-mutated or deficient ccRCC lines
and pa- tient-derived xenografts.45 PT2399 had greater
activity than sunitinib, was active in sunitinib-progressing
tumors, and was better tolerated45 (Figure 2).
The first-in-class clinical HIF-2a inhibitor PT-2385
caused dramatic tumor regressions in patient-derived